Naegleria fowleri is a free-living amoeba found in freshwater, soil, hot springs, and poorly chlorinated swimming pools [1]. In humans, N. fowleri trophozoites cause primary amoebic meningoencephalitis by penetrating the nasal mucosa and migrating to the brain via the olfactory nerves [1,2]. Naegleria can induce acute and severe tissue inflammatory responses associated with lytic necrosis [2], and N. fowleri can cause host cell death in both contact-dependent and contact-independent manners [2,3]. It has also been reported that N. fowleri induces reactive oxygen species–dependent necroptosis in host cells [4]. Interestingly, N. fowleri did not induce the cleavage of caspase 3 of Jurkat T cells in co-culture system [4]. Although host cell death is an important aspect of pathogenesis in infection with N. fowleri, the detailed mechanisms involved in N. fowleri–induced host cell death are still not fully understood.
O-linked β-N-acetylglucosamine (O-GlcNAc) glycosylation (O-GlcNAcylation) is the main form of glycosylation in the cytosolic and nuclear compartments of eukaryotic cells [5]. Similar to phosphorylation, O-GlcNAcylation is a reversible post-translational modification in which O-GlcNAc is added to serine or threonine residues of nuclear and cytoplasmic proteins [5,6]. O-GlcNAcylation of proteins is also linked to glucose metabolism because O-GlcNAcylation is an end point of the hexosamine biosynthetic pathway [8,10]. In mammals, O-GlcNAcylation is regulated by only 2 enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), which catalyze the addition and removal of O-GlcNAc, respectively [5,6]. O-GlcNAcylation regulates various cellular processes such as apoptosis, cell proliferation, transcription, translation, and signal transduction [5]. An increase in O-GlcNAcylation is associated with cell proliferation and survival, whereas a decrease (O-deGlcNAcylation) is related to cell cycle arrest and cell death [6–8]. Recently, it was reported that Entamoeba histolytica induces O-deGlcNAcylation of proteins in HepG2 cells during cell death [9]. However, the signaling role of O-GlcNAcylation in host cell death induced by N. fowleri is unknown. In this study, we investigated whether the modulation of O-GlcNAcylation is an important signaling event in N. fowleri–induced Jurkat T cell death.
N. fowleri trophozoites (Carter NF69; ATCC No. 30215) were cultured under axenic conditions in Nelson’s medium at 37°C. Jurkat T cells (clone E6-1; ATCC No. TIB-152TM) were grown in RPMI 1640 medium (Gibco/Invitrogen, Gaithersburg, MD, USA) with 10% fetal bovine serum (Gibco/Invitrogen) in a humidified 5% CO2 incubator at 37°C.
To observe N. fowleri–induced cell death caused by DNA fragmentation, Jurkat T cells (4×106 cells/sample) were incubated with N. fowleri trophozoites at a ratio of 1:2 or 1:1 (Jurkat T cells to N. fowleri) for 3 or 6 hr at 37°C in a humidified CO2 incubator. After incubation, the cells were harvested by centrifugation and washed with cold phosphate-buffered saline (PBS). DNA was extracted using a TaKaRa kit (MK600; Shiga, Japan) according to the manufacturer’s protocol. DNA samples were separated by electrophoresis on a 2% agarose gel and visualized using ethidium bromide to assess DNA fragmentation. For LDH release assay, Jurkat T cells (2×105 cells/sample) pretreated with or without PUGNAc were incubated with live N. fowleri for 3 hr at a 1:1 ratio (Jurkat T cells to N. fowleri). After incubation, LDH release was determined by evaluating the amounts of LDH in culture supernatants using the CytoTox 96 cytotoxicity assay system (Promega, Madison, WI, USA) according to manufacturer protocol. The background (spontaneous LDH release) value was measured in non-stimulated cells and subtracted from each measurement. Maximum LDH release was measured by incubating non-stimulated cells in a lysis solution (1% Triton X-100 and PBS) at 37°C for 45 min. For immunoblotting, Jurkat T cells (5×105) were pretreated with or without O-(2-Acetamido-2-deoxy-D-glucopyranosylidenamino) N-phenylcarbamate (PUGNAc) (Toronto Research Chemicals, North York, Canada) for 4 hr and stimulated for the indicated time periods in the presence or absence of N. fowleri. After incubation, cells were lysed using lysis buffer (50 mM Tris-HCl, pH 8.0; 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM ethylenediaminetetraacetic acid, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, and protease inhibitor cocktail) on ice for 30 min. Cell lysates were centrifuged at 15,000 g for 5 min, loaded onto an SDS-PAGE gel, and transferred onto a polyvinylidene fluoride membrane (Millipore, Billerica, Massachusetts, USA), followed by blocking with 5% skim milk. The blot was probed with primary antibodies at 4°C overnight, followed by incubation with appropriate secondary horseradish peroxidase–conjugated antibodies. Immunoreactivity was detected using LumiGLO (Cell Signaling Technology, Danvers, Massachusetts, USA). Mouse anti–O-GlcNAc and rabbit anti–β-actin monoclonal antibodies were purchased from Abcam (Cambridge, UK) and Cell Signaling Technology (Beverly, Massachusetts, USA), respectively.
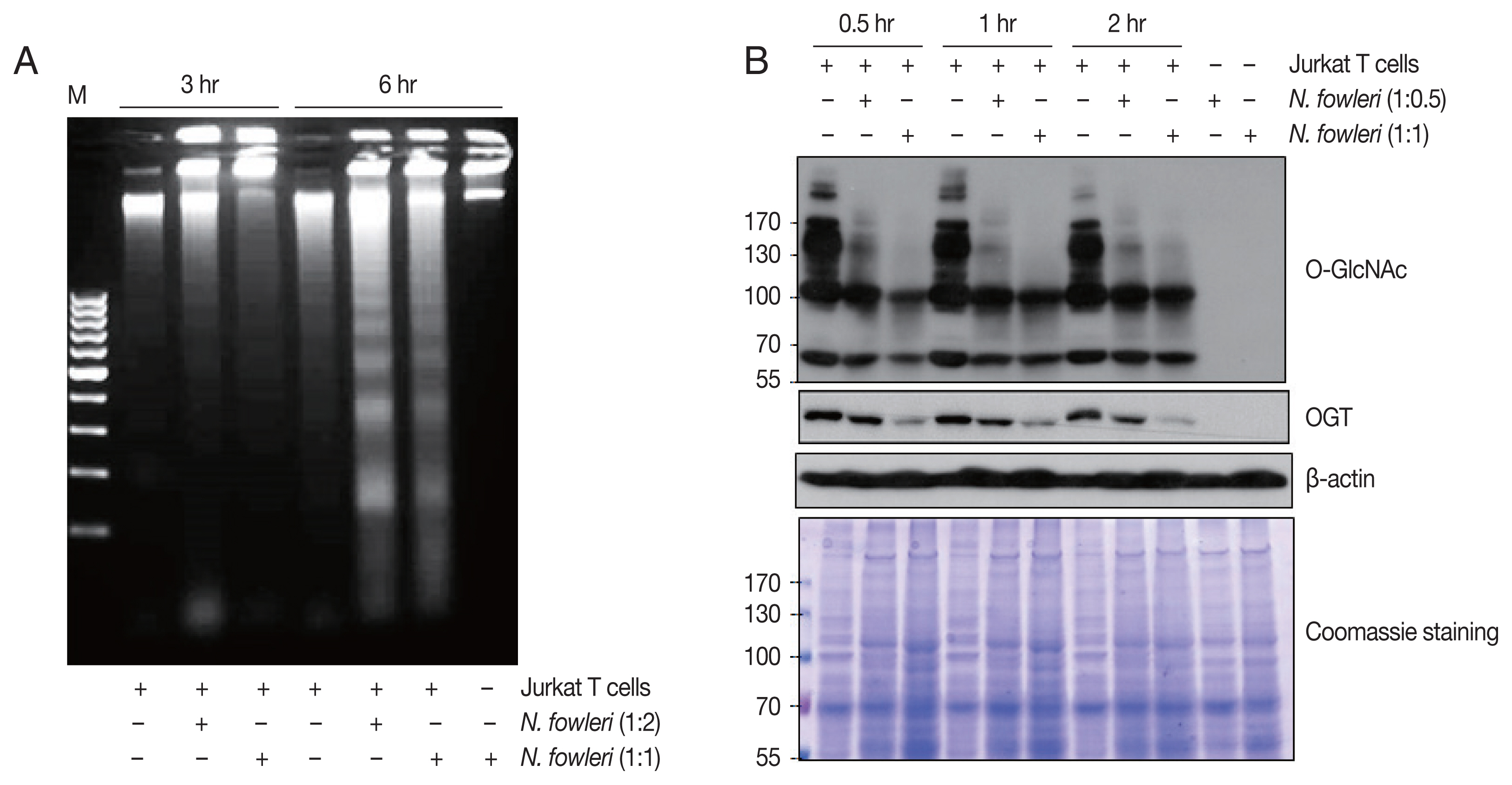
Co-incubation with N. fowleri trophozoites induced cell death and O-deGlcNAcylation in Jurkat T cells. As shown in Fig. 1A, N. fowleri–induced DNA fragmentation was observed in cells incubated at a ratio of 1:2 or 1:1 (Jurkat T cells to N. fowleri) for 3 or 6 hr. DNA fragmentation occurred in a time- and dose-dependent manner; co-incubation of Jurkat T cells with amoebae for 6 hr at a ratio of 1:2 induced robust DNA fragmentation.
Next, we investigated whether co-incubation with N. fowleri induced the modulation of O-GlcNAcylated proteins. SDS-PAGE results (Fig. 1B) showed that patterns of protein bands of various sizes were almost equivalent between cells incubated with or without N. fowleri. However, the results of immunoblotting with anti–O-GlcNAc antibody indicated a marked decrease in the level of O-GlcNAcylated proteins in Jurkat T cells incubated for 0.5 hr with N. fowleri at a ratio of 1:0.5 or 1:1 (Jurkat T cells to N. fowleri), compared to the level in cells incubated with medium alone. In particular, high-MW (>100 kDa) O-GlcNAcylated proteins disappeared from cells in the presence of N. fowleri, in a manner that was dependent on the amoeba load. In contrast, when N. fowleri alone was incubated with anti–O-GlcNAc antibody, in the absence of Jurkat T cells, no reacted protein bands were observed.
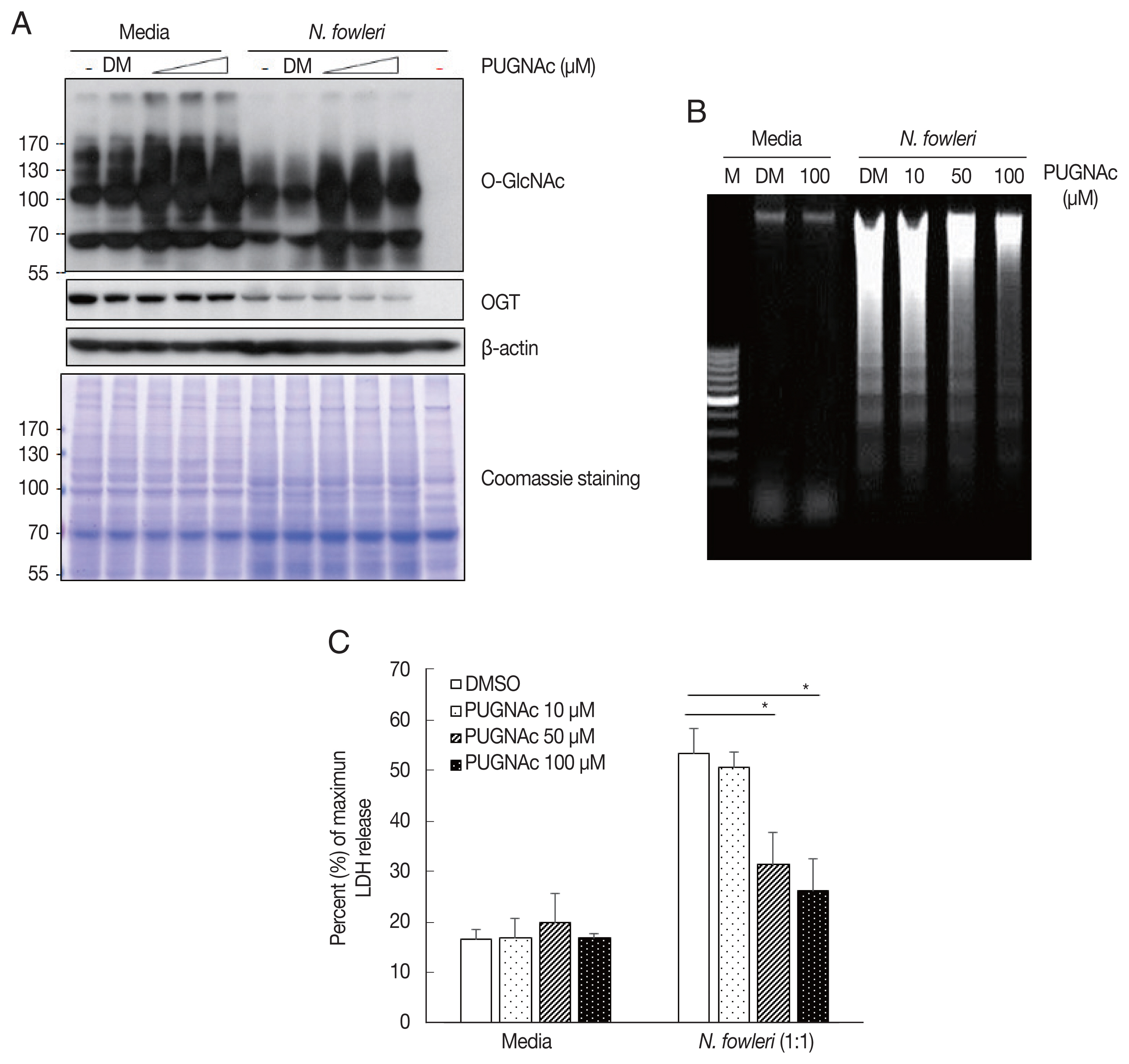
To examine whether OGA is involved in N. fowleri–induced O-deGlcNAcylation, we used the OGA inhibitor PUGNAc to prevent the activity of OGA in Jurkat T cells. As shown in Fig. 2A, the level of O-GlcNAcylated protein increased in resting-state Jurkat T cells pretreated with PUGNAc for 4 hr, compared to that in the vehicle control. In addition, pretreatment with PUGNAc prevented N. fowleri–induced O-deGlcNAcylation.
We next investigated whether OGA inhibition using PUGNAc affects Jurkat T cell death induced by N. fowleri. As shown in Fig. 2B, pretreatment with 50 μM or 100 μM PUGNAc inhibited N. fowleri–induced DNA fragmentation compared to the control. Similarly, pretreatment of host cells with PUGNAc also inhibited N. fowleri-induced LDH release (50 μM: 31.5± 6.2%; 100 μM: 26.1±6.5%) compared to results for cells pretreated with DMSO (53.5±4.7%) (Fig. 2C). These results suggest that O-deGlcNAcylation is closely related to the N. fowleri–induced Jurkat T cell death process.
Homeostasis between O-GlcNAcylation and O-deGlcNAcylation in mammalian cells is maintained through the balance of OGT and OGA activity [5,6]. For example, an increase in the level of O-GlcNAcylated protein is associated with increased OGT activity or decreased OGA activity. Conversely, a decrease in the level of O-GlcNAcylated protein is associated with decreased OGT expression or increased OGA expression [6,8,10]. Here, we showed that N. fowleri caused a decrease in the O-GlcNAcylated protein level in Jurkat T cells in a dose-dependent manner within 30 min. The OGT expression in Jurkat T cells was gradually reduced upon co-incubation with N. fowleri (Fig.1A). This result is consistent with the recent report that protozoan parasite E. histolytica induces O-deGlcNAcylation by decreasing OGT expression during host cell death. However, the expression level of OGA of host cells was relatively not changed in Entamoeba-treated HepG2 cells [9]. Therefore, we investigated the signaling mechanism responsible for N. fowleri–induced O-deGlcNAcylation using the OGA inhibitor PUGNAc. Pretreatment of Jurkat cells with PUGNAc prevented the decrease in the level of O-GlcNAcylated proteins, which was consistent with a recent report of an E. histolytica–induced host cell death model [9]. These results suggest that imbalance between OGT and OGA activity is highly involved in N. fowleri–induced O-deGlcNAcylation in Jurkat T cells.
The alteration of intracellular O-GlcNAcylated protein levels by cellular stimuli/stress is related to cell fate because O-GlcNAcylated proteins are tightly regulated to maintain homeostasis in the resting state [11]. Increased levels of O-GlcNAcylated proteins are associated with cell proliferation, survival, and activation, whereas decreased protein levels are associated with cell death, damage, and deactivation [6–8]. Several studies have reported that an increase in O-GlcNAcylated protein levels protects against cell death and various cellular stresses. For example, the addition of glucosamine leads to increased OGT expression, thereby increasing cellular resistance to stimulation and decreasing cell death rates [6,7]. Additionally, inhibition of OGA activity using a pharmacological OGA inhibitor, such as streptozotocin or PUGNAc, attenuates the cell death process [12,13]. According to a recent report, human T lymphoblastic cells treated with PUGNAc showed significant reductions in tributyltin-induced apoptosis [14]. In addition, Entamoeba-induced cell death was significantly prevented by pretreatment with an OGA inhibitor [9].
In conclusion, we demonstrated for the first time that N. fowleri–induced O-deGlcNAcylation in Jurkat T cells contributes to host cell death. Our study demonstrates a novel analysis of the mechanism underlying N. fowleri–induced host cell death, which may be helpful in elucidating the pathogenesis of N. fowleri infections in humans.